paired end sequencing r1 and r2
The map is 80 but the R1 R2 counts are. As indicated in the comments yes you can definitely tell standard Illumina sequencers to sequence mates in a pair to different lengths.
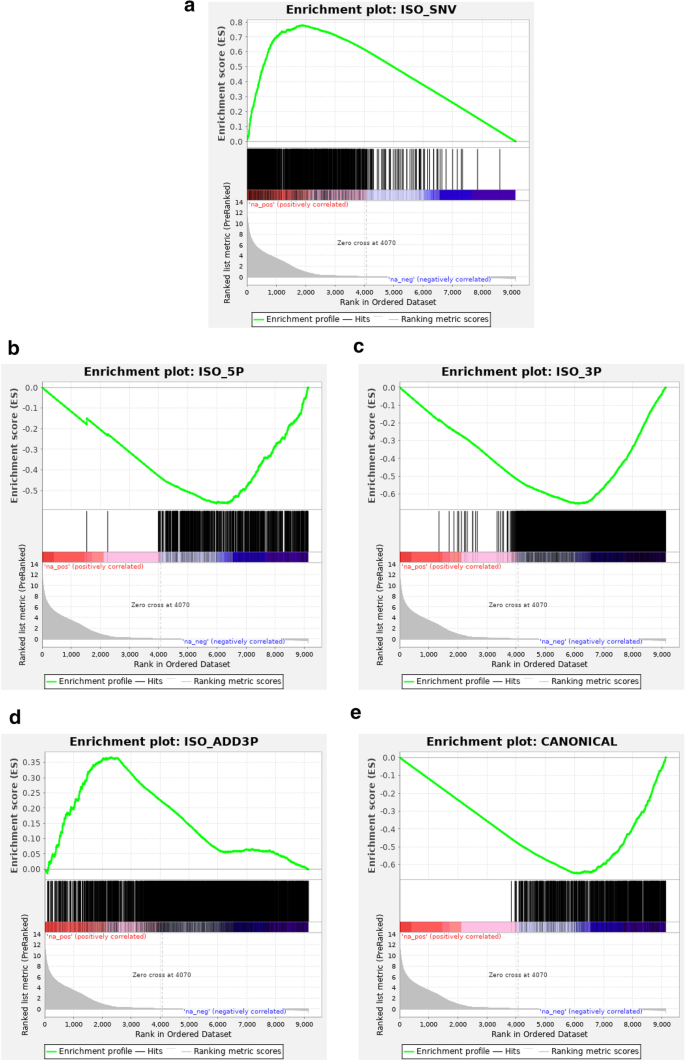
Paired End Small Rna Sequencing Reveals A Possible Overestimation In The Isomir Sequence Repertoire Previously Reported From Conventional Single Read Data Analysis Bmc Bioinformatics Full Text
You could see the reverse.
. First sequencing can either be single-end where each sample has only sequence data or paired-end where each sample has two sequence data R1 and R2. The steps below can be. For the first test I took some sequence from the human genome hg19 and created two 100 bp reads from this region.
The files have this naming convention. The reads have been generally trimmed to remove the low quality reads and adaptor contaminations. This usually does not happen for PE sequencing which makes me a bit nervous to proceed with analysis.
The inner mate distance. There are four fastq files and are named like this. For the paired-end mode we initially joined reads using.
Are paired end reads reverse. For each dataset we used the miRNA module using paired-end mode and single end mode for the R1 and R2 reads. If the sequencing sample is the same as the original copy the read R1 should be mappable in.
10-09-2013 1013 AM. I have some data files in fastq format generating from paired -end sequencing. Now lets get started.
For each cluster that passes filter a single sequence is written to the corresponding samples R1 FASTQ file and for a paired-end run a single sequence is also. If the sequencing sample is the same as the original copy the read R1 should be mappable in forward direction in 5-end and R2 in reverse direction in 3-end. The Illumina paired-end sequencing technology can generate reads from both ends of target DNA fragments which can subsequently be merged to increase the.
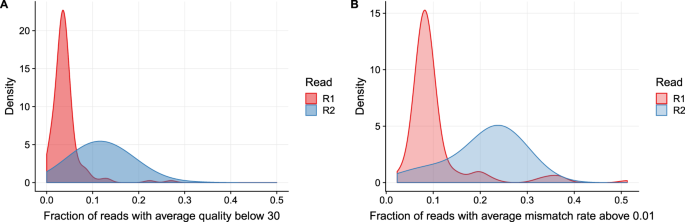
2 shows the proportion of low quality R2 reads additional base mismatches in the R2 read relative to the R1 read is correlated with the amount of fragments. There are two FastQ files generated in an Illumina paired-end reads sequencing run. Download scientific diagram The direction and positional order of the paired-end reads R1R2.
This is quite common in single-cell RNA-seq. For paired-end alignment aligners want the R1 and R2 fastq files to be in the same name order and be the same length. Adapter trimming can remove FASTQ sequences if.
If youre running MiSeq Reporter 23 theres a new setting called StitchReads that can do this for you. So while R1 starts with your 5 adapter for R2 anything is possible depending on the length of the fragment of interest and the read length. I received paired-end illumina data for some DNA sequencing Hiseq.
Simply add StitchReads 1 to your Settings.
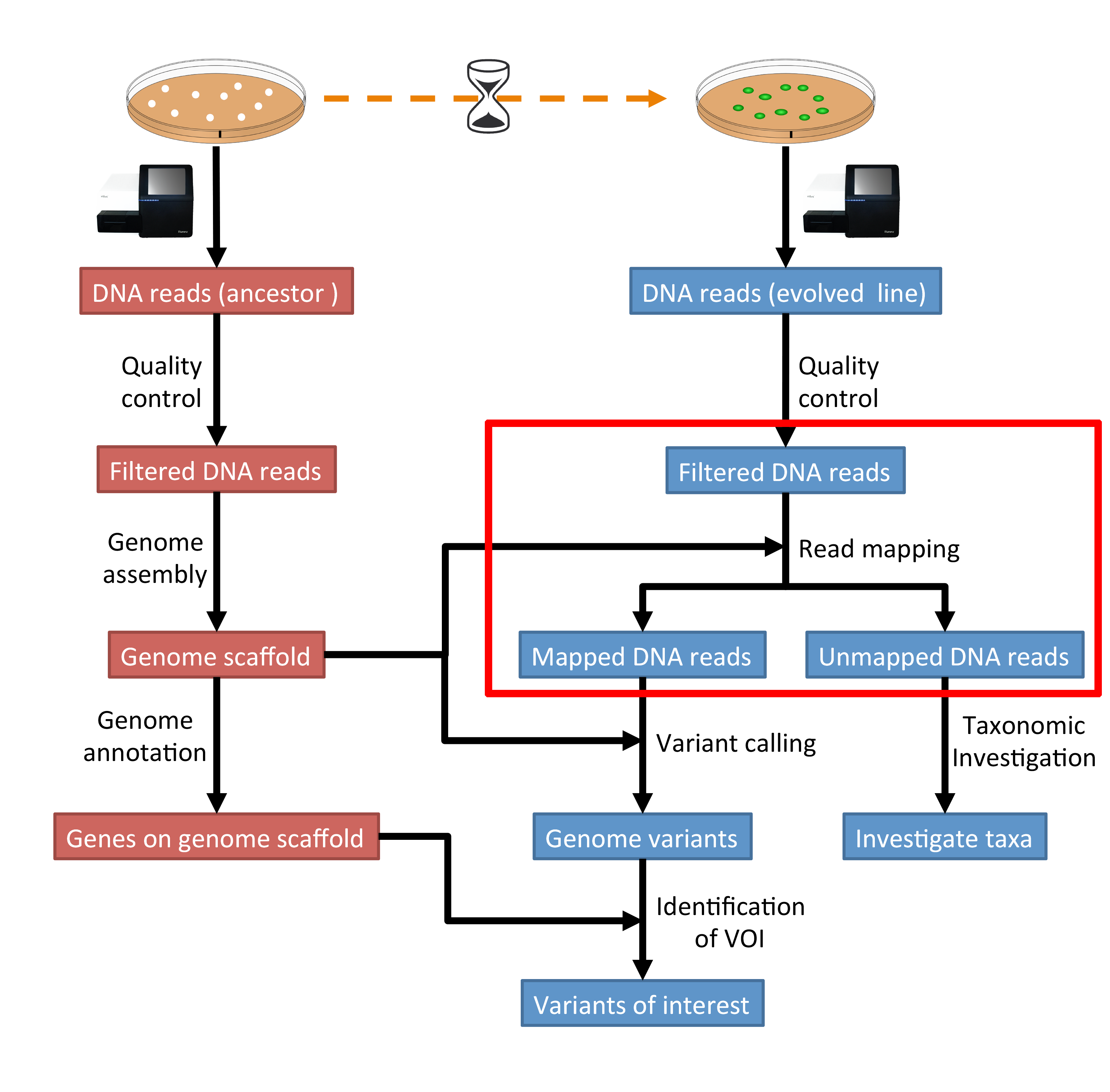
5 Read Mapping Genomics Tutorial 2020 2 0 Documentation

Casava Files Of Paired End Reads General Discussion Qiime 2 Forum

Quality Assessment Of Raw Fastq Files Of 100 Bp Paired End Reads R1 Is Download Scientific Diagram

Dmx With 4 Initial Read Files I1 I2 R1 R2 User Support Qiime 2 Forum

Figure 1 Iguide Method For Crispr Off Target Detection Springerlink
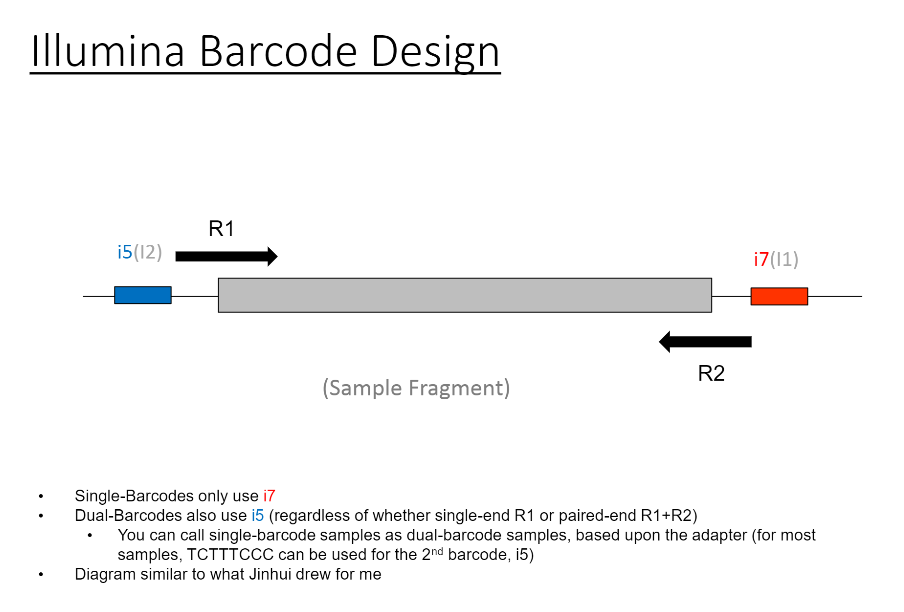
Calling Single Barcode Samples From Mixed Runs As Dual Barcode Samples Possible Illumina Run Qc Flags

Trouble With Merging Paired End Reads User Support Qiime 2 Forum

Deal With Single End And Paired End Data Bioinformatics

Radseq Why Do Reverse Radtags Have Different Start Points In Radtag Sequencing Bioinformatics Stack Exchange
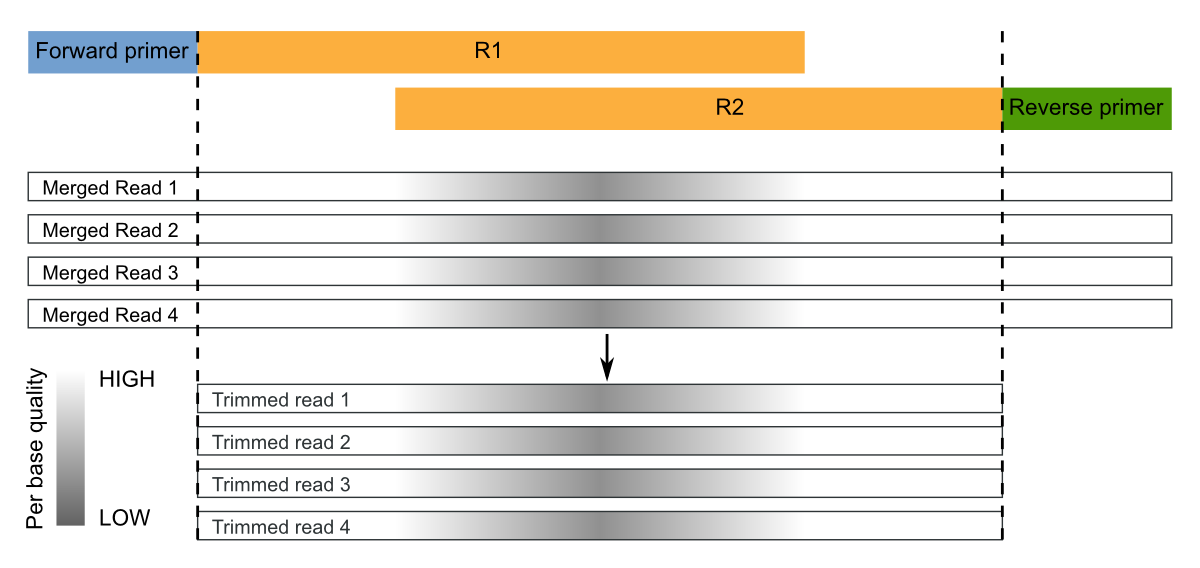
Paired End Sequencing Micca 1 6 2 Documentation
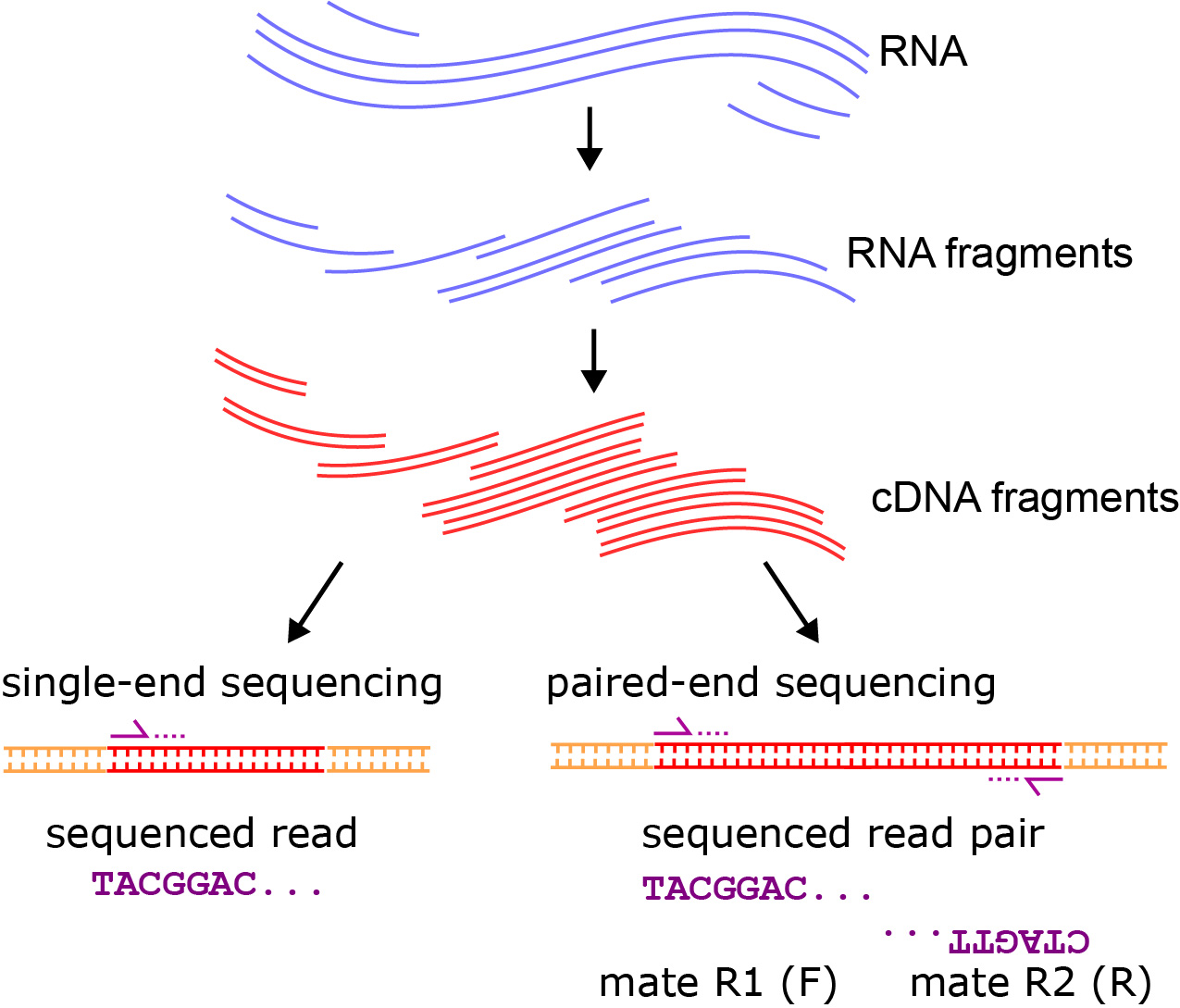
Chapter 6 Transcriptomics Applied Bioinformatics
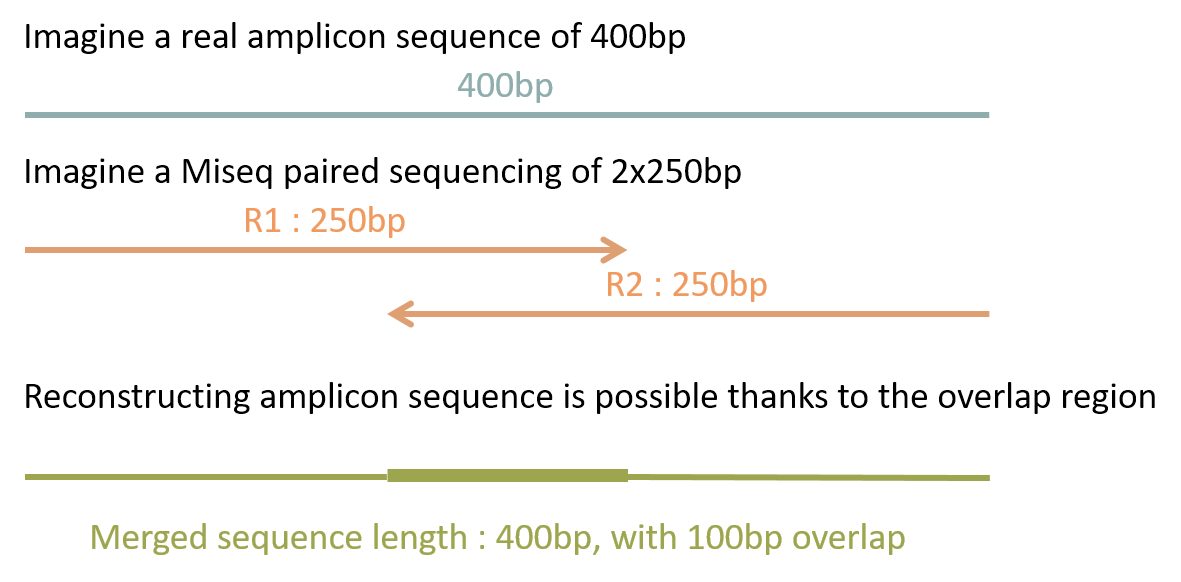
Galaxy

The Direction And Positional Order Of The Paired End Reads R1 R2 If Download Scientific Diagram
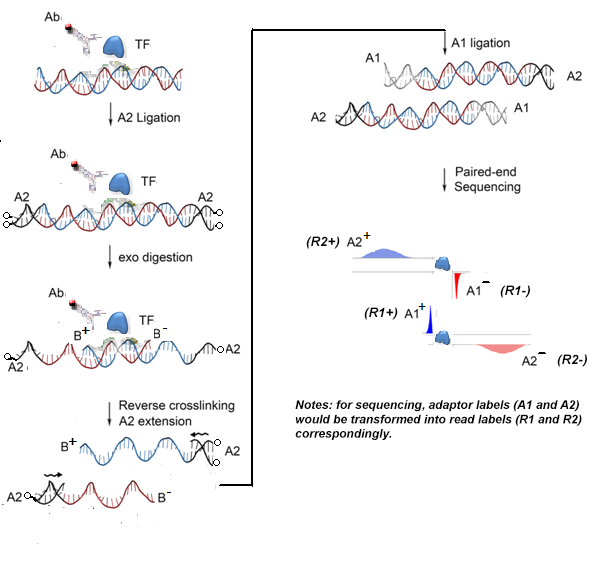
Introduction Epest Chip Exo Paired End Sequencing Processing Toolkit 1 0 Documentation

Detection Of Rearrangement By A Paired End Reads And B Junction Download Scientific Diagram

How I Will Import Combined R1 Fastq And R2 Fastq Files Into Qiime2 User Support Qiime 2 Forum
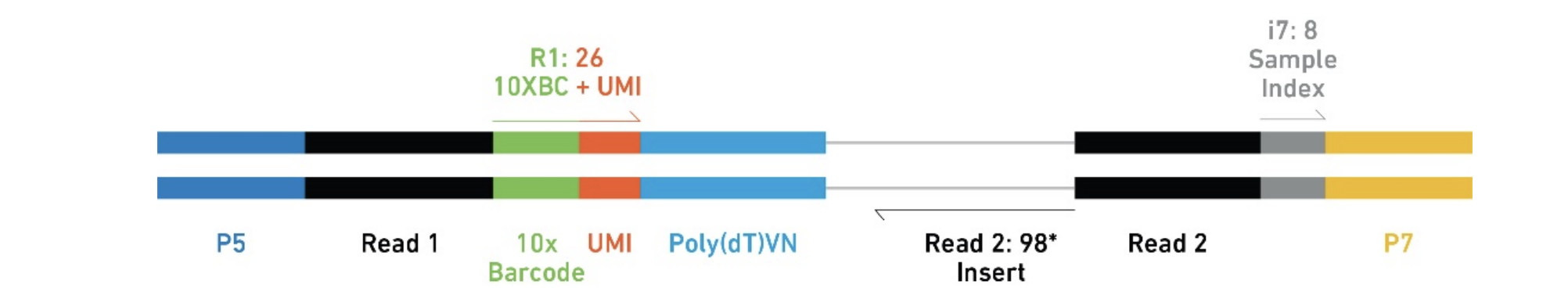
Understand 10x Scrnaseq And Scatac Fastqs Dna Confesses Data Speak

Reconstruction Of Full Antibody Sequences In Ngs Datasets And Accurate Vl Vh Coupling By Cluster Coordinate Matching Of Non Overlapping Reads Sciencedirect
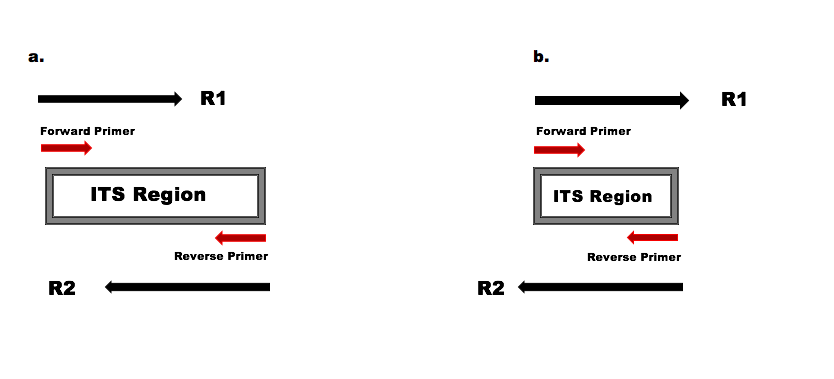
Dada2 Its Pipeline Workflow 1 8